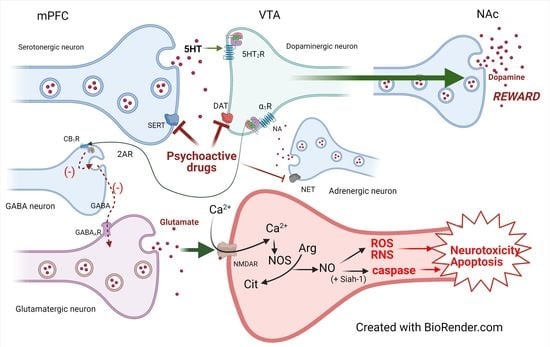
Рисунок 3. Предполагаемые общие механизмы стимулирования дофаминергической нейротрансмиссии катинонами. Катиноны ингибируют обратный захват серотонина (5-HT) и дофамина (DA) из синаптической щели, блокируя активность транспортеров 5-HT (SERT) и DA (DAT), что впоследствии приводит к накоплению 5-HT, который стимулирует постсинаптические 5-HT2A - рецепторы (5-HT2R ) , способствующие высвобождению ДА в синаптической щели (и который затем остается на этом уровне и не может быть повторно захвачен). Создано на
BioRender.com (по состоянию на 22 января 2021 г.).
Исследования на животных показывают, что мефедрон стимулирует двигательные функции, а его постоянное употребление вызывает прогрессирующую потерю 5-НТ и DA-нейронов в гиппокампе и полосатом теле [
111 ,
112 ,
113 ]. Причина такого массивного повреждения DA-нейронов еще не выяснена, но предполагается, что основной причиной может быть окислительный стресс, вызванный АФК и РНС. Происхождением этих реактивных видов являются нейротрансмиттеры, особенно DA, и влияние психоактивных веществ на митохондриальные ETC [
111 ,
114 ,
115 ,
116 ,
117 ,
118 ,
119 ]. Несмотря на антиоксидантные системы, свободные радикалы накапливаются, вызывая немедленное молекулярное повреждение (например, карбонилирование белков, перекисное окисление липидов) [
17 ] и деградацию клеточных органов, играя важную роль в развитии и прогрессировании нейродегенеративных заболеваний [
111 ]. У подростков окислительный стресс, вызванный злоупотреблением психоактивными препаратами, влияет как на кортикальные нейроны, так и на его передачу в подкорковые структуры, что приводит к последствиям для когнитивных функций [
120 ,
121 ]. В целом употребление психостимулирующих препаратов оказывает прооксидантное действие и в гиппокампе и префронтальной коре [
113 ] снижает общую антиоксидантную способность, активность антиоксидантных ферментов, а также увеличивает концентрацию малонового диальдегида (МДА) [
122 ] ,
123 ], поскольку в мозге содержится значительное количество липидов и переходных металлов, что приводит к нарушению межнейронной передачи [
121 ,
122 ]. Среди клинических проявлений, появляющихся после употребления веществ со структурой β-кетоамфетамина, выделяют гипокинезию и дистонию, предполагающие изменения экстрапирамидной системы, аналогичные болезни Паркинсона [
124 ] с оговоркой , что применение мефедрона не вызывают тремор покоя [
79 ]. Возможная причина этих симптомов — накопление марганца [
125 ], используемого при синтезе препарата [
126 ], во внутреннем бледном шаре [
127 ] и в компактной части черной субстанции [
128 ], где он оказывает различное цитотоксические эффекты, включая образование свободных радикалов и апоптоз в полосатом теле [
129 ,
130]]. Противоядия от интоксикации марганцем или мефедроном не существует, а классические противопаркинсонические препараты при этом синдроме неэффективны [
79 ,
131 ].
3.7. Производные амфетамина
В случае употребления амфетамина и метамфетамина, подобно мефедрону, немедленные эффекты вызваны вмешательством в передачу нейронов DA [
132 ]. Эти вещества проникают в нейрон и вызывают массивный выброс нейромедиатора в синаптическую щель. Исследования на грызунах показывают, что амфетамины повышают уровни маркеров окислительного стресса, таких как MDA, SOD, глутатион (GSH/GSSG), 2,3-дигидроксибензойная кислота, в коре головного мозга, полосатом теле [
133 ] и гиппокампе [
134 ]. Более того, токсические дозы метамфетамина ингибируют ЦЭТ, воздействуя на все четыре комплекса, в полосатом теле , гиппокампе , миндалевидном теле, хвостатом ядре и префронтальной коре , что инкриминируется в развитии нейродегенеративных заболеваний [
135 ,
136 ,
137 ]. Гибель нейронов в этих областях происходит в результате апоптоза [
138 ] вследствие изменения соотношения между проапоптотическим (Bax, Bad) и антиапоптотическим белками (Bcl-2, Bcl-XL), что приводит к активации каспаз 9 и 3. [
139 ,
140 ]. Нейротоксичность, вызванная амфетамином и/или кокаином [
141 ], опосредуется глутаматергической системой (
рис. 4 ), активацией рецепторов NMDA [
139 ,
142 ] после внутрицитоплазматического притока Ca 2+ [
133 ,
143 ,
144 ] и активации НОС. Вслед за повышением концентрации НА в синаптической щели будет наблюдаться повышение концентрации ГЛУ в нейронах мПФК (угнетение тормозного контроля высвобождения нейромедиатора гамма-аминомасляной кислотой, ГАМК). Этому увеличению можно способствовать, например, путем ингибирования обратного захвата НА кокаином. Следовательно, NA активирует α1-адренорецепторы, расположенные в нейронах DA VTA. Этот процесс способствует высвобождению 2-арахидонилглицерина (2-АР) в синаптическую щель, тем самым активируя эндогенный каннабиноидный путь. Липидный медиатор действует на постсинаптические каннабиноидные рецепторы (CB1R), предотвращая высвобождение ГАМК. Таким образом, отрицательный контроль приостанавливается и происходит повышенное высвобождение GLU [
145 ]. Благодаря активации NOS после притока Ca 2+ новообразованное соединение может генерировать свободные радикалы, особенно пероксинитрит [
146 ], который взаимодействует с гидроксильным радикалом, образующимся по реакции Габера-Вейсса/Фентона [
137 ,
147 ,
148 ,
149].]. Помимо сосудорасширяющего эффекта после активации цГМФ, NO может нитрозилировать белки, которые модулируют процесс апоптоза. Одним из таких белков является глицеральдегид-3-фосфатдегидрогеназа (GAPDH), которая поступает в ядро в нитрозилированной форме и способна взаимодействовать с Siah1 [
150 ], как показано на
рисунке 5 .